
KT-474 - a case study in PROTAC clinical translation - so far, so good
7th December 2023

Most early PROTAC programmes focused on oncology. Early efforts on BRD4 proved to have poor tolerability but good traction around well-known targets such as androgen receptor, estrogen receptor and BTK quickly allowed projects to move to the clinic to test bifunctional TPD agents with the first drug approvals potentially now within reach.
Indeed, many new drug modalities are initially focused in oncology as, although the area is extremely crowded, there remains significant unmet need with opportunities for accelerated approval and, in some cases, patients who may tolerate a less clean drug safety profile due to the severity of their disease.
It’s for these reasons that many viewed Kymera’s choice of IRAK4 for autoimmune diseases as their lead programme as a bold decision. The target itself had good genetic validation (and healthy human knockout individuals to infer likely good safety) but small molecule IRAK4 inhibitors had previously come up short in trials so there were questions around target validity. Now for protein degradation approaches, this should be a good scenario - where genetic inactivation would be expected to give efficacy but simple inhibition doesn’t recapitulate this, suggesting target knockdown is needed – but previous clinical failures by target modulators have a habit of souring investor sentiment and interest in the target which then needs to be overcome. Coupled to this that the expectation that a very good safety profile would be needed to give an attractive treatment option in patients with non-life threatening conditions such as atopic dermatitis, and the path to approval for a new PROTAC modality vs IRAK4 looked challenging.
However, the team at Kymera have now successfully shown preliminary safety and efficacy data in patient cohorts with their lead IRAK4 degrader KT-474, representing a significant milestone for the TPD field, namely showing the potential for application, and ultimate approval, for non-oncology indications.
Their recent manuscript in Nature Medicine is a really good, concise summary of the pre-clinical and clinical profile of KT-474 and brings out some of the potential advantages (but also some of the unknowns) of degrader approaches over traditional small molecule inhibitors.
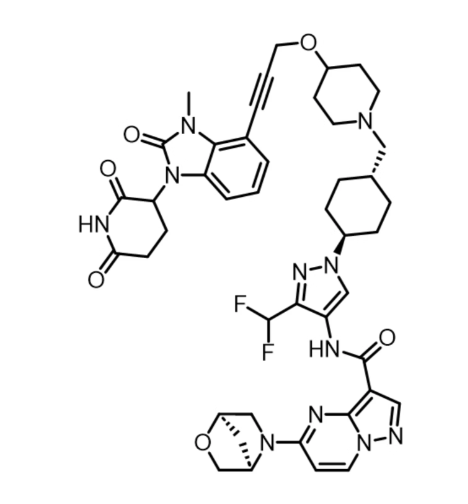
The structure of KT-474 itself (Below), follows the now well established design brief of linking a first generation cereblon binder to a derivative of a previously disclosed series of IRAK4 inhibitors through carefully optimised exit vectors. The linker, so often critical to getting the right pharmacokinetic & efficacy profile, features just 5 rotatable bonds as well as a weakly basic centre to give an appropriate balance of solubility, oral bioavailability and metabolic stability.
As would be expected from using a fairly selective IRAK4-binding moiety, proteomic selectivity of degradation is high though interestingly, other members of the MYDDosome (eg IRAK1, MyD88) do not appear to be destabilised upon removal of IRAK4 from the complex in this system. Careful design of KT-474 also abrogated any cereblon-mediated, molecular glue-like degradation or IKZF1/3 & SALL4, known activities of the IMiD class of drugs linked to cell survival or teratogenicity effects, clearly undesirable in this class of medicines.
In an area where IRAK4 inhibitors have underperformed in clinical settings, differentiation over historic inhibitors is a crucial step to give confidence that a degrader approach would give different clinical outcomes. In head-to-head comparisons with IRAK4 inhibitor PF-06650833 in PBMCs, KT-474 showed only modestly improved inhibition of IL-6 or IL-8 secretion following TLR4 or TLR7 stimulation, though when assessing TLR9-mediated pathway activation (NF-kB activation by CpG-B in B cells), IRAK4 degradation showed an effect whereas inhibition did not. The IRAK4 degrader clearly then had some functional advantages over target inhibition in cellular systems though whether this would translate into a meaningful clinical advantage was perhaps not assured from these data.
The encouraging pre-clinical profile was however sufficient to give confidence to advance KT-474 to clinical studies.
Clinical tolerability across the Feb 2021-Oct 2022 phase 1 study was generally good with low levels of AEs noted. Early observations of modest, asymptomatic QTc prolongation, an effect which may have led to some concern in these patient populations and amongst investors, diminished over time and was seen to be fully reversible.
The human pharmacokinetic profile of KT-474 is a good example of that seen with many PROTAC degraders, namely dose proportional increase in exposure (up to 1000mg dose) with low inter-subject variability and a long half-life leading to modest accumulation on repeat dosing with low plasma clearance and high levels of tissue penetration leading to skin:plasma ratios >10 in many cases. Indeed, the low clearance resulted in sustained skin drug exposure for at least 2 weeks after cessation of dosing indicative of low metabolic clearance or diffusion out of tissues.
Upon single dose treatment, plasma drug was largely cleared by 7 days post-dose whilst IRAK4 degradation in PBMCs was maintained at >70% at this time point in almost all doses above the 25mg starting dose suggesting slow resynthesis of IRAK4 (or else sustained degradation driven by more sustained drug exposure in PBMCs relative to plasma).
Repeat dosing gave the anticipated increase in degradation with all doses (25mg-200mg) showing >90% IRAK4 degradation in PBMCs after qd dosing for 14 days. The effect in skin however, was somewhat weaker (despite much higher drug exposure in this tissue) with doses of 50mg and above showing a maximal reduction in IRAK4 or ~70% and requiring up to 14 days for maximal effect. The reasons for this weaker effect in skin are unclear though could be due to higher tissue binding leading to lower free drug levels or else the observation that certain cell types, including skin can show lower sensitivity to CRBN-based degraders, due potentially to lower CRBN expression.
Levels of IRAK4 degradation in patients were comparable to healthy volunteers with a ~50% reduction or IRAK4 in patient skin sufficient to reduce IRAK4 levels to those seen in healthy volunteers, though not considerably lower.
KT-474-induced reductions in IRAK4 levels were accompanied by functional benefit, namely inhibition of TLR4 or TLR7 signalling as shown using ex vivo assays on patient blood however, this effect would also be expected with an IRAK4 inhibitor so the degree of functional advantage from IRAK4 degradation cannot easily be estimated.
Clinical evaluation (see eg here or here) of IRAK4 inhibitor PF-06650833 in healthy volunteers also showed a decrease in acute phase responses including in CRP and a panel of inflammation-relevant genes though the effect was modest. Where comparison could be made inhibitor and degrader showed similar effects suggesting these markers do not optimally capture potential advantages of IRAK4 degradation.
Crucially, patients dosed with KT-474 also showed clinical improvements as measured using a range of scales (assessing lesions, itch & pain amongst others), with effects potentially more significant than seen with an IRAK4 inhibitor previously (where comparison was possible). This represents an exciting initial efficacy datapoint, with further studies now ongoing.
Overall, the KT-474 dataset are a very instructive example of the potential power of TPD therapies in a clinical setting and show the opportunity to deliver genuine patient benefit. Stepping back, from a TPD-centric perspective, it’s difficult to see a clear “night and day” inflammatory pathway functional advantage of an IRAK4 degrader relative to an IRAK4 inhibitor from the data presented, however, clear patient benefit has been seen with the degrader in the small patient cohorts assessed here, giving a foundation for more extensive studies. This response was also accompanied with a good clinical tolerability profile, underlining that the PROTAC class of molecules is unlikely to carry any class-specific concerns.
The clinical pharmacokinetic and pharmacodynamic profiles also show great encouragement for the field. Given KT-474 lies well outside traditional (“rule of 5”) areas of chemical property space, the well-behaved clinical pharmacokinetics and oral dosing show that good molecular design can overcome any perceived issues using larger molecules. In vivo biodistribution and pharmacodynamic response remains an area of intense study in the TPD field and KT-474s profile serves to highlight some of the opportunities and issues. While response in terms of IRAK4 degradation and functional pathway inhibition in the blood compartment (PBMCs) is rapid and sustained, this profile cannot necessarily be directly extrapolated to tissues: KT-474 is seen to penetrate skin (and potentially other tissues) at high levels though IRAK4 lowering appeared to be slower and less complete compared to blood. Once in skin, clearance was also slower suggesting a “slow in, slow out” tissue penetration profile which may provide opportunities though also some challenges compared to dosing protocols and PK-PD models developed for traditional “fast in, fast out” small molecules. Understanding drug tissue binding in a more systematic way or else using inspiration from biologic drug development models may help here.
Now these issues of differential efficacy in different tissues and compartments are not unique to TPD drugs, they are likely present with many other drugs also. TPD drugs however have an inbuild biomarker, namely monitoring the protein levels of the target. This means it is easier to identify subtle biodistribution effects. The more you look, the more you see. This also makes TPD agents valuable tools for understanding whole body and clinical pharmacological action in a higher level of detail than is possible with many other approaches.
This KT-474 study is a valuable example of a detailed analysis of activity of a new TPD agent – it underscores the potential of the TPD field, the potential of IRAK4 degraders to treat inflammatory disease and, perhaps most importantly in the longer term, the potential for TPD drugs to increase our understanding of whole body clinical pharmacology to a new level.
Kudos to the Kymera team for generating the data but then also for taking the time to summarise it in such an accessible format. Thanks guys.

© 2023-2024
All rights reserved. Janus Drug Discovery Consulting Ltd
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.