
When DEL met TPD - a match made in heaven?
4th November 2023
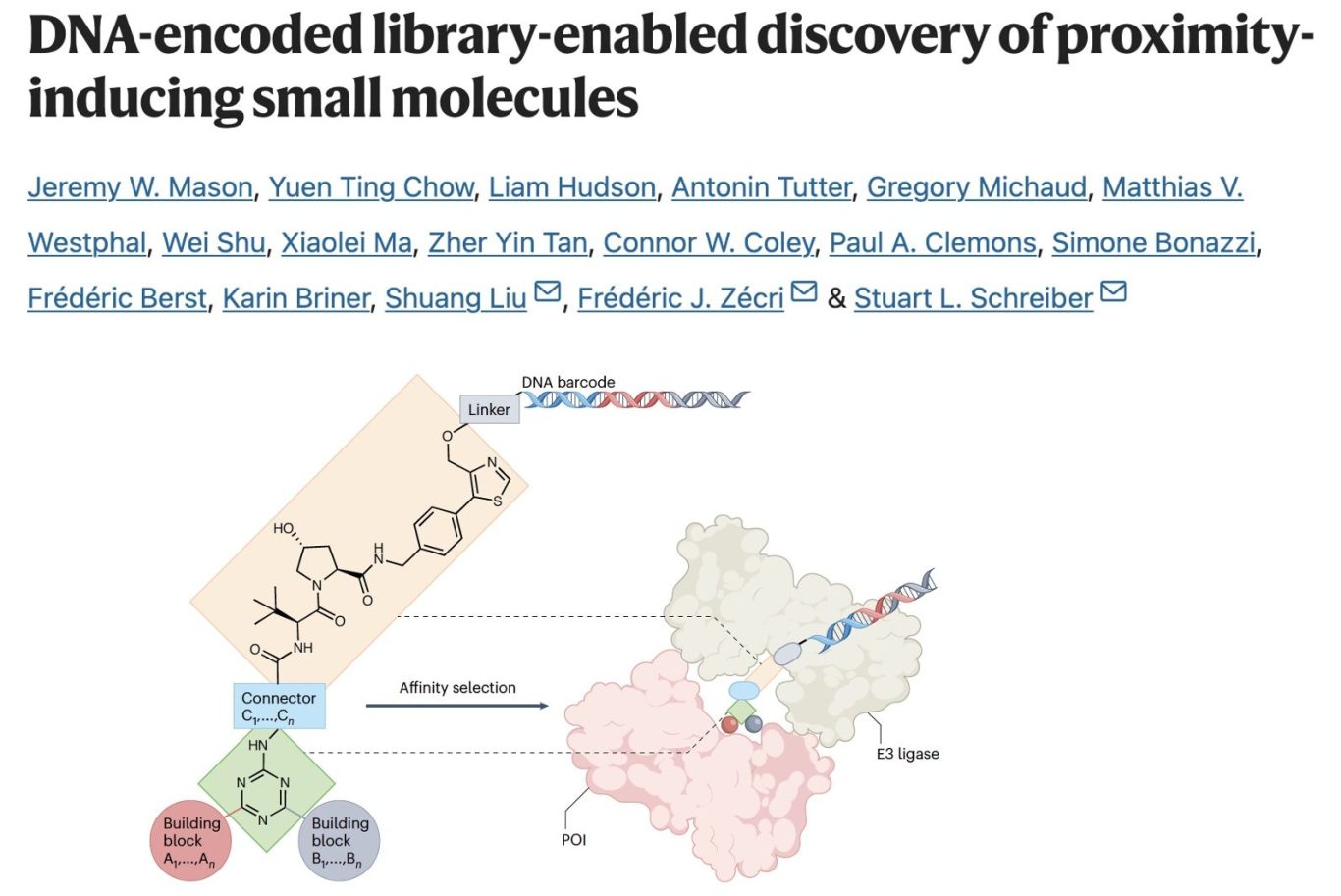
DNA-encoded library (DEL)-screening has been around for a long time now and allows the simultaneous screening of millions of molecules (or billions to trillions if you really want to - though it has catches) to find binders to proteins. The molecules which are identified from this affinity-based screening using DNA bar codes still bear a linker (to the DNA) which needs to be subsequently removed but this linker also conveniently defines an accessible exit vector from the protein binding site so it quickly became an enticing route to apply to TPD and PROTAC discovery.
It all seemed to make sense even in the early days of TPD back in 2012: Take your favourite undruggable target and perform a DEL screen to find binders. You will also find all those lovely non-functional binders which are good for TPD applications but traditional, function-based inhibitor screening will miss. The binders you find come with a pre-defined exit vector so all you need to do is use it to introduce a linker then a ligand to your favourite E3 ligase (VHL, CRBN etc) and job done. Degradation of any protein in the proteome in a game-changing advance for drug discovery.
Except it hasn't quite delivered in that way, yet.
The trouble with undruggable targets is that it is rather hard to find binders to them (they are undruggable after all, or more relevantly, unligandable) and DEL screens are no more likely to find binders to difficult targets than other forms of high throughput screens (though fragment screens will likely give better hit rates and a chance to optimise to a higher affinity binder). And, even when you have a target binder with enough affinity (1uM may be ok though <10nM makes things a whole lot easier), you still need to use it to make a degrader which will form the right ternary complex etc - not a given in all cases. Molecular glues may access some of these hard-to-ligand targets where binary complexes are impossible but ternary ones can be stable however.
So, with these various factors, unfortunately the literature is not yet replete with PROTACs to undruggables discovered through a conveyer belt of DEL screens. Maybe all pharma companies are sitting on a stack of great DEL-PROTAC stories just waiting to see the light of day as the resultant drugs progress through the clinic, but I'd perhaps be surprised.
Wind forward to today and the recent publication in Nature Chemical Biology from Stuart Schreiber and others (See also a nice precis from Andrea Testa here) who have a different approach.
With TPD, function is king, which is why screening directly for degraders of your protein of interest (POI) using HiBit, ELISA, Western blot etc is the most common approach to find them. There are simply so many factors you need to get right to see degradation (cell uptake, binary binding, ternary complex binding, ubiquitin transfer rate, overcoming protein resynthesis or DUB action etc etc) that if you focus on just one of these in a reductionist approach, you might find yourself focused on not the most challenging part of the problem. Instead you should screen for what is closest to the degradation endpoint as possible.
With previous DEL-TPD applications, the DEL screen sought to find a binary binder to the POI. A challenging step yes, but still very distal to the function of actually showing target degradation. Using DEL to find degraders directly has not yet proven possible due to the pooled nature of the DEL libraries but screening to find agents able to induce a ternary complex is certainly a step in the right, functional direction and that is what the authors here have done.
Using good old-fashioned triazine chemistry to create a large library of potential POI binders, they prepared a library of 1M+ VHL-based PROTACs. This was possible making use of a quirk of VHL that it has 2 accessible exit vectors so you can link on the DNA barcode (through the "triazole" exit vector) with reduced risk that this linker will foul up the ternary complex between VHL and the POI. This is very useful but unfortunately a trick it is much more difficult to pull with CRBN and likely most other E3 ligases.
With this library in hand, it enables screening for ligands which induce ternary complexes (by affinity screening POI in presence and absence of VHL). Perhaps unsurprisingly, BRD4 was selected as a probe target. Certainly not undruggable but a good sandbox, even if it may not be representative of other, more challenging and clinically-relevant targets. The DEL screens identified a range of binary binders and ternary complex facilitators. Crucially, the strength of ternary complex formation was seen to predict depth of degradation - a correlation often seen with VHL-based PROTACs made using non-DEL routes, but often less clear with CRBN-based agents.
So, a DEL-based approach which moves beyond simply finding binary binders and now can identify molecules able to form ternary complexes which subsequently appear to be good degraders.
Will the avalanche of discovery of degraders of undruggable targets now begin? A step in the right direction but it may yet be a long road.

© 2023-2024
All rights reserved. Janus Drug Discovery Consulting Ltd
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.