
I feel the need...the need for speed
TPD drugs are catalytic. So what?
20th November 2024
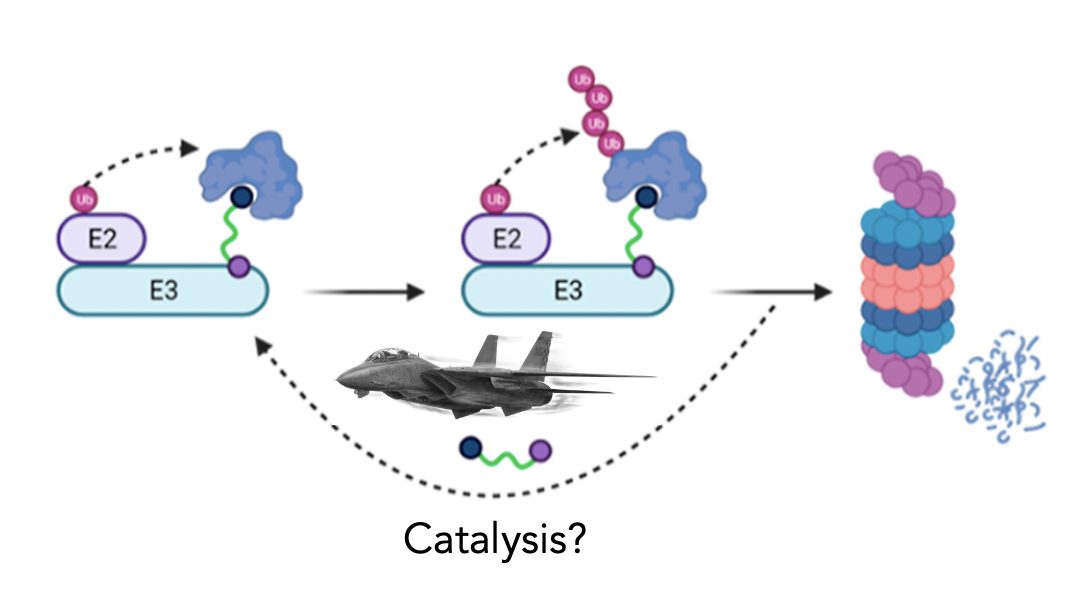
One of the key advantages often cited for TPD drugs is their ability to utilize event-driven pharmacology. In other words, a single molecule of TPD drug, whether it’s a glue or bifunctional PROTAC, can mediate the degradation of multiple molecules of target in a departure from the “occupancy-driven” pharmacology which has underpinned drug discovery for well over 100 years.
So TPD drugs are catalytic. So what?
Drugs using event-driven pharmacology are also termed “catalytic” or “sub-stoichiometric”, a characteristic which stems from the fundamental concept of induced-proximity TPD drugs which facilitate/stabilise/agonise a substrate:E3 ligase interaction to kick off a one-way ubiquitylation cascade and target destruction, usually in the proteasome. Once the TPD drug has introduced the substrate to the ligase to initiate this cascade, its job is done and so it can then go and find another substrate protein molecule and do it all again, and again, and again. Hence the catalysis.
All of this means then that in theory you should require fewer molecules of TPD drug to deal with a given population of target protein molecules (ie protein of interest; POI) which could translate to lower doses of drug being effective in the clinic, But does this catalysis matter in the real world? Evolution has figured out the advantages of using event-driven pharmacology – imagine if enzymes weren’t catalytic and were single-use machines, not a great recipe for life.
Most groups cite catalysis as the reason their TPD drugs work with higher potencies (often much higher than the binary binding Kd of the drug to its target) but few ever dig into this to quantify the effects. The Abbvie group however have taken a good look at this in a quantitative case study of some CRBN-based BTK PROTACs published recently and it’s a great read. NB The study used PROTACs though, in principle, molecular glues should adhere to the same framework – after all PROTACs and glues are just the same but on different parts of the ternary complex co-operativity continuum.
To derive a numerical measure of catalytic efficiency (ie turnover number – how many protein molecules could a single PROTAC molecule send for degradation), they needed to carry out some careful measurements of i) the starting intracellular concentrations of POI & E3 complex ii) the first order kinetic rate constant of degradation and iii) the intracellular free concentration of TPD drug.
Now, factors i) and ii) are easier to derive via quantitative proteomics and a typical HiBiT kinetic degradation profile respectively though the intracellular free drug is trickier. The authors chose to calculate this using Kpu,u ratios of free drug in cell & media but sometimes highly-bound PROTACs can give unreliable results in these assays – also much of the useful PROTAC may actually be bound to POI and/or E3 so the “free” conc may not be the only variable to consider (as the authors acknowledge) – so there are caveats but overall, I think the conclusions they see are valid.
Fortunately, the Abbvie team handled the maths so we don’t need to, but a few interesting observations came out of their elegant work.
Straight away, the concentration of BTK in cells (360nM) was around ~6 fold higher than CRBN. This baseline POI:E3 (“degradee to degrader”) ratio is important. More E3 ligase relative to POI means the E3 (and potentially the TPD drug) may have to work less hard to degrade a given POI population than a scenario where the POI may be in huge excess with only a small amount of E3 to do the job. We see this in practical terms where some cells have much lower CRBN levels than others and can show much slower degradation and weaker DC50 values as the low CRBN conc. struggles to chew through a swimming pool of POI.
If we then take a look at the absolute numbers in more detail we see that, in a single TMD8 cell, there are around 300,000 BTK protein molecules but, in cells treated with 5nM of PROTAC “D1”, this only equates to around 100 molecules of free PROTAC per cell (taking around an hour for this concentration to be reached – confirming a “slow in” profile).
The PROTAC is clearly outnumbered and to deplete all cellular BTK, each PROTAC molecule must tackle around 3000 BTK protein molecules each (to keep things simple, the authors use cycloheximide-treated cells to prevent the cell busily producing de novo synthesised BTK while the PROTAC is trying to remove it).
Indeed, the PROTAC can deplete >90% BTK within 7h so, when the numbers are crunched, this leads to an initial rate of ~1500 molecules of BTK degraded per hour per PROTAC molecule. Ie each PROTAC facilitates the degradation of a substrate protein molecule once every ~2 seconds – ie it is pretty efficient.
Now, these numbers look impressive (a related PROTAC, MT-802 was also good though the catalytic rate was ~5 fold lower) – indeed at very short time points (<1h) where the intracellular free PROTAC level is still increasing due to slow cell uptake, rates of >10,000 BTK/h/PROTAC (ie one PROTAC degrading one BTK protein every 0.3s) are inferred.
Let’s also not forget that D1 may not have been a highly optimised PROTAC – even better efficiency is likely possible with PROTACs that have come from good med chem work.
Also emerging from these numbers is the fact that to mediate a high level of degradation, much less than 1% of the cellular population of CRBN is engaged. This leaves over 99% of CRBN to carry on with its normal functions giving a low chance of undesired effects on physiological CRBN substrates. Even at much higher PROTAC doses, it’s still likely much CRBN remains available.
The authors also highlight some other interesting observations. PROTAC D1 is actually not a selective BTK degrader. It uses a promiscuous kinase binder so it’s no surprise that it actually degrades many kinases (as shown by usual proteomics methods) but when the authors elegantly looked at catalytic rates of degradation of many of these kinases they saw that BTK was uniquely fast (AURA/B, PTK2B, ITK, LCK, LYN were also pretty good) and that there appeared to be a broad relationship between catalytic rate and extent of degradation (though an imperfect relationship, perhaps influenced by protein expression level & resynthesis rate). Fast is good.
What does all this mean? Can we use it to design better degraders? Perhaps.
Catalysis clearly is important here – without it, it’s unlikely PROTACs would give any useful level of cell or in vivo activity which could support drug development. Whatever values you pick for the intracellular drug concentration (with the complex caveats the authors outline), the molecules are clearly mediating turnover of multiple POI molecules without the cell really noticing that CRBN function has been hijacked in any meaningful way. If your POI is high abundance (BTK is highly expressed in B cells where you want to degrade it), this catalysis is probably rather important – if the POI is lower expression (or the POI:E3 ratio is tipped in your favour), it may be less important. As POIs often have different expression levels in different cells, the importance of catalysis can be complex to assess.
The authors go on to try to dissect what factors may underlie the different catalytic rates. Rate doesn’t seem to rely on the binary affinity of the PROTAC for BTK or CRBN (which we probably already knew) and it also seems that more stable/co-operative ternary complexes are likely better (which we probably already knew) but there are many factors at play here. There are many opportunities to get high levels of catalysis and degradation and, at the end of the day, that is what we want and need.
Faster degraders are usually better degraders – unless they’re also rapidly degrading unwanted targets of course. And, once you go in vivo, intuitively, faster also seems like it should be better. Your drug only has a finite time in the body to do its thing before it is metabolized or cleared so it needs to make the best use of that time and chew through as many POI molecules as it can before its gone.
The Abbvie team have done an amazing job here, kudos to them, and I’m so glad they did the hard work to put some real numbers behind the “event-driven” narrative but they’ve needed to generate a lot of data to get to these conclusions, data that you may not have the time or resource to generate on your degrader project.
They highlight a range of factors which can be used to optimise the catalytic rate of degradation but measuring these parameters is often tricker than running a high throughput (eg HiBiT) degradation assay. Most projects will (or at least should) use this cellular degradation assay as the workhorse but remember, kinetics are king in the TPD world. Running a degradation assay at 24h often doesn’t tell you much about kinetics as many compounds start to reach the same endpoint after extended periods. Run your DC50 at 4h or 2h or even 1h if you can – that will highlight the really fast ones which are probably the better ones.
Tom Cruise is not often cited in the scientific literature but he was clearly giving TPD scientists sage advice: “I feel the need… the need for speed”.

© 2023-2024
All rights reserved. Janus Drug Discovery Consulting Ltd
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.